

Sickle cell anaemia - B. Pharma 2nd Semester Pathophysiology notes pdf
Sickle cell anaemia
Content
• Sickle cell anaemia
• Pathogenesis
• Clinical manifestation
• Treatment
Objective
At the end of this PDF Notes, student will be able to
• Explain Sickle cell anaemia
• Describe the pathogenesis of Sickle Cell Anaemia and Clinical features of it
• Treatment of sickle cell anaemia
Sickle Cell Anaemia
• Sickle-cell disease (SCD), or sickle-cell anaemia or drepanocytosis, is an autosomal co-dominant genetic blood disorder characterized by red blood cell that assume an abnormal, rigid, sickle shape.
Haemoglobinopathies: Haemoglobin in RBCs may be abnormally synthesised due to inherited defects. These disorders may be of two types:
• Qualitative disorders e.g. sickle cell syndrome, other haemoglobinopathies.
• Quantitative disorders e.g. thalassaemias.
Sickle syndromes occur in 3 different forms:
• 1. As heterozygous state for HbS: sickle cell trait (AS).
• 2. As homozygous state for HbS: sickle cell anaemia (SS).
• 3. As double heterozygous states e.g. sickle β-thalassaemia, sickle-C disease (SC), sickle-D disease (SD).
• Sickle cell anaemia (SS) is a homozygous state of HbS in the red cells in which an abnormal gene is inherited from each parent.
• Red blood cells typically live 90–120 days, but sickle cells only survive 10–20 days
Pathogenesis of
1. Basic molecular lesion: single point mutation in one amino acid out — there is substitution of valine for glutamic acid –
- 6-residue position of the β-globin, producing Hb α2β2s2.
2. Mechanism of sickling: During deoxygenation, the red cells containing HbS change from biconcave disc shape to an elongated crescent-shaped or sickle-shaped cell- sickling
• Form elongated rod-like polymers
• Which align and distort the red cell into classic sickle shape
3. Reversible-irreversible sickling
4. Factors determining rate of sickling:
• i) Presence of non-HbS haemoglobins
• ii) Intracellular concentration of HbS.
• iii) Total haemoglobin concentration.
• iv) Extent of deoxygenation.
• v) Acidosis and dehydration.
• vi) Increased concentration of 2, 3-BPG in the red cells.
Sickle Cell Anemia vs. Sickle Cell Trait
• People who have sickle cell anemia are born with it; means inherited, lifelong condition.
• They inherit two copies of sickle cell gene, one from each parent.
• Sickle cell trait is different from sickle cell anemia. People with sickle cell trait don’t have the condition, but they have one of the genes that cause the condition.
• People with sickle cell anemia and sickle cell trait can pass the gene on when they have children.
Inheritance of Sickle Cell Anemia
•
• None will have sickle cell anemia.
• The parent who has sickle cell anemia (HbSS) can only pass the sickle hemoglobin gene to each of their children.
• If both parents have sickle cell trait (HbAS) there is a one in four (25%) chance that any given child could be born with sickle cell anemia.
• There is also a one in four chance that any given child could be completely unaffected.
• There is a one in two (50%) chance that any given child will get the sickle cell trait.

Clinical Features of Sickle Cell Anemia
• Painful episodes
• Pneumococcal disease
• Acute chest syndrome
•
• Splenic sequestration
• Stroke
• Osteonecrosis
• Priapism
• Retinopathy
• Leg ulcers
• Gallstones
• Renal abnormalities
• Osteopenia
• Nutritional deficiencies
• Placental insufficiency
• Pulmonary hypertension

| Associated with higher hemoglobin | Associated with lower hemoglobin |
| Painful episodes | Stroke |
| Acute chest syndrome | Priapism |
| Osteonecrosis | Leg Ulcers |
| Proliferative retinopathy |
Complications of Sickle Cell Disease

Sickle Cell – Avascular Necrosis

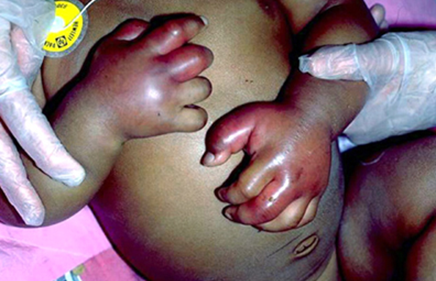
Sickle Cell – Dactylitis
Sickle Cell Anemia – Treatment
• Opiates and hydration for painful crises
• Pneumococcal vaccination
• Retinal surveillance
• Transfusion for serious manifestations (eg stroke); exchange transfusion
• Hydroxyurea
• Stem cell transplant
Treatment
— Effective treatments are available to help relieve the symptoms and complications of sickle cell anemia, but in most cases there’s no cure.
— The goal is to relieve the pain; prevent infections, eye damage, strokes and control complications if they occur.
— Pain medicine: acetaminophen, nonsteroidal anti-inflammatory drugs (NSAIDs), and narcotics such as meperidine, morphine, oxycodone, and etc.
— Heating pads
— Hydroxyurea, Folic Acid
— Blood Transfusions
Prevention of
— Identify what can trigger the “Crisis” such as stress, avoid extremes of heat and cold weather, don’t travel airplane that is not cabin pressurized
— Maintain healthy lifestyle habits
÷ Eating healthy
÷ Avoid dehydration
÷ Exercise regularly
÷ Get enough sleep and rest
÷ Avoid alcohol and don’t smoke
— Regular medical checkups and treatment are important
Summary
• Sickle-cell disease (SCD), or sickle-cell anaemia or drepanocytosis, is an autosomal co-dominant genetic blood disorder characterized by red blood cell that assume an abnormal, rigid, sickle shape.
• Pathogenesis: Basic molecular lesion, Mechanism of sickling, Reversible-irreversible sickling ,Factors determining rate of sickling
• Sickle cell trait is different from sickle cell anemia. People with sickle cell trait don’t have the condition, but they have one of the genes that cause the condition

0 Comments: